
Differential Analysis in Genomic Data Science - R
Summary
Differential analysis in Genomic Data Science involves comparing gene expression or other genomic features between different experimental conditions. Genomic data, often obtained through technologies like RNA sequencing, undergoes preprocessing and normalization to ensure accuracy. Statistical methods are then applied to identify genes or genomic regions that exhibit significant differences in expression, with multiple testing corrections used to minimize false positives. The results are visualized to highlight significant changes, providing insights into the biological processes underlying different conditions such as diseases or treatments. This analysis is crucial for understanding how the genome responds to various factors and revealing key genes or pathways involved in specific biological contexts.
This project aims to investigate a genomic data science file. This file includes expression data for KIRC (kidney renal clear cell carcinoma) patients where some patients did not receive any treatment (control) while others received a treatment (treatment). In the expression data, the first 20 columns correspond to “control” samples while the next 20 to “treatment” samples. An analysis is performed to identify the genes that are differentially expressed between these two groups and identify the genes that are significant at p<0.01 after multiplicity correction (Benjamini-Hochberg).
Load Library
library(glmnet)
library(tibble)
library(dplyr)
library("DESeq2")
library("pheatmap")
library(MASS)Preprocessing Data
DeSeq2 allows for computation of High-throughput count data but needs an annotation file. Hence, we create the annotation file based on the colnames of the kirc_count. The design matrix will be based on that column from the annotation file:
# read the file
kirc_counts <- read.csv("ExpressionData.csv", row.names = 1)
#create new data frame for annotation
kirc_annotation <- data.frame(condition = rep(c("control", "treated"), each = 20))
rownames(kirc_annotation) <- colnames(kirc_counts)
#The annotation file should look as:
head(kirc_annotation,5)# verifying if true
all(rownames(kirc_annotation) == colnames(kirc_counts))## [1] TRUEFinally using DESeqDataSetFromMatrix:
#Turn the condition to a factor
kirc_annotation$condition <- factor(kirc_annotation$condition)
#Using DESeq
kirc <- DESeqDataSetFromMatrix(countData = kirc_counts,
colData = kirc_annotation,
design = ~ condition)
kirc## class: DESeqDataSet
## dim: 4990 40
## metadata(1): version
## assays(1): counts
## rownames(4990): A2BP1|54715 AAAS|8086 ... ZW10|9183 ZXDB|158586
## rowData names(0):
## colnames(40): C1 C2 ... T19 T20
## colData names(1): conditionAlso, let us consider the “control” as the reference category.
kirc$condition <- relevel(kirc$condition, ref = "control")Differential expresssion analysis
After these preparations, we are now ready to jump straight into differential expression analysis. Our aim is to identify genes that are differentially expressed between the treated and the controlled cells using a single function, DESeq.
kirc_deseq = DESeq(kirc)Results tables are generated using the function results, which extracts a results table with log2 fold changes, p-values and adjusted p-values:
res = results(kirc_deseq)
head(res[order(res$padj),], 3)## log2 fold change (MLE): condition treated vs control
## Wald test p-value: condition treated vs control
## DataFrame with 3 rows and 6 columns
## baseMean log2FoldChange lfcSE stat pvalue
## <numeric> <numeric> <numeric> <numeric> <numeric>
## CDKN2A|1029 146.295 5.63703 0.377905 14.9165 2.57415e-50
## TNFAIP6|7130 1721.861 7.36261 0.493238 14.9271 2.19643e-50
## FABP6|2172 315.642 7.70519 0.566845 13.5931 4.39847e-42
## padj
## <numeric>
## CDKN2A|1029 6.02994e-47
## TNFAIP6|7130 6.02994e-47
## FABP6|2172 6.86894e-39We can check the number of differential expressed genes significant at adjusted p-value cutoff of 0.01.
cat("The number of genes that are differential express:",sum(res$padj < 0.01, na.rm=TRUE)) ## The number of genes that are differential express: 435Extraction and Visualization for selected genes
The next objective is to Extract the expression data for the selected genes:
#filter to get the deferentially expressed genes
kicr_diff_exp_genes <- subset(res, padj < .01 )
#verify the number of values is correct from part (a)
nr <- nrow(kicr_diff_exp_genes)
#Some of the the names of the deferentially expressed genes
head <- head(rownames(kicr_diff_exp_genes),10)
cat("The number of rows:", nr, "\n")## The number of rows: 435cat("Some names of the differentially expressed genes:", head, "\n")## Some names of the differentially expressed genes: AASS|10157 AATF|26574 ABI2|10152 ABI3|51225 ACN9|57001 ACO1|48 ACOT12|134526 ACOT1|641371 ACTR10|55860 ADH1A|124Next, we extract the expression data of the selected genes from the original data set. The rlogTransformation function, used later, avoids returning matrices with NA values, in the case of a row of all zeros, by returning zeros (essentially adding a pseudocount of 1 only to these rows). However, we will replaced the values by 1 to all zero counts beforehand before calling the function rlogTransformation.
kicr_selected_genes <- subset(kirc_counts, rownames(kirc_counts) %in% rownames(kicr_diff_exp_genes))
#Any 0 count is added by a small number 1
kicr_selected_genes[kicr_selected_genes == 0] <- 1#Rerun Deseq on the new subset selected genes
kicr_selected_genes_DeSeq2 <- DESeqDataSetFromMatrix(countData = kicr_selected_genes,
colData = kirc_annotation,
design = ~ condition)
kicr_selected_genes_DeSeq2$condition <- relevel(kicr_selected_genes_DeSeq2$condition, ref = "control")
kirc_deseq3 = DESeq(kicr_selected_genes_DeSeq2)
#rlogTransform the counts
kicr_rlog2 = rlogTransformation(kirc_deseq3)
#plotPCA(pas_rlog2, intgroup=c("condition"))
select = order(rowMeans(assay(kicr_rlog2)), decreasing = TRUE)
# show_rames do
pheatmap(assay(kicr_rlog2)[select, ],scale = "row",
show_rownames = F, annotation_col = kirc_annotation)
By using heatmap on all selected genes, we can be clearly seen as two distinguish groups on the top of the chart, which is similar to the actual groups (treated and control). Likewise, we can see a distinct division into two large groups row wise. In total, we can make up about 4 quadrants in the heat map.
Pathway Analysis
Pathway analysis in Genomic Data Science involves the systematic examination of biological pathways to understand how genes work together in specific cellular processes. It begins with the identification of genes that exhibit significant changes in expression or other genomic features through methods like differential analysis. These identified genes are then mapped onto predefined biological pathways, such as those from databases like KEGG or Reactome. Statistical methods are applied to assess the enrichment of these genes in particular pathways, helping researchers infer the functional significance of the genomic alterations. Pathway analysis is valuable for gaining insights into the broader biological context of genomic data, uncovering the relationships between genes, and elucidating the molecular mechanisms underlying different conditions or experimental treatments.
The next step is to perform a pathway analysis using the diferentially expressed genes. Note that the gene names in the expression data include two types of IDs separated by a “|”. Hence, we need to extract only the 1st part, for example, for a gene ID “XCR1|2829” you need to extract “XCR1”
# extract only the first part
rownames(kicr_selected_genes) = sub("\\|.*", "", rownames(kicr_selected_genes))
# Verify the substring is extracted
head(rownames(kicr_selected_genes), 40)## [1] "AASS" "AATF" "ABI2" "ABI3" "ACN9" "ACO1"
## [7] "ACOT12" "ACOT1" "ACTR10" "ADH1A" "ADORA3" "AGAP4"
## [13] "AGFG2" "AKAP4" "ALPK2" "AMT" "ANKLE2" "ANXA2P1"
## [19] "ANXA8" "AP2A1" "APLNR" "ARHGAP11A" "ARHGAP11B" "ARHGAP27"
## [25] "ARHGAP33" "ARHGAP9" "ARID3B" "ARRDC2" "ASB9" "ATAD5"
## [31] "ATP12A" "ATP13A5" "AVEN" "BACH1" "BATF" "BAZ1B"
## [37] "BHLHA15" "BPI" "BSPRY" "BTN2A1"# output the file to be inputted at david.ncifcrf.gov
write.table(rownames(kicr_selected_genes), "inputtoDavid.txt",
row.names = FALSE,
col.names = FALSE,
quote = FALSE)Steps used in David:
Inputted as the text file
Converted into Official_Gene_Symbol after selecting “Not Sure” Indentifier
We look into the KEGG pathways.
Below is the enrichment result for the KEGG pathways, with EASE of .05 and at least 2 pathways genes in a list. There are 6 pathways that are significant.
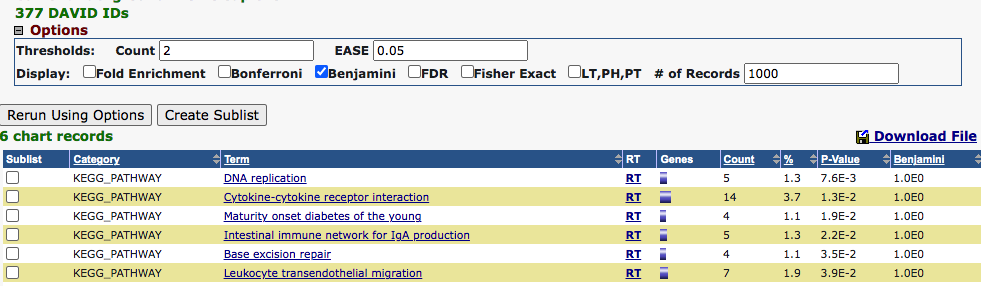
It seems to indicate that KIRC may have an effect on cell proliferation and glucose uptake.
Prediction.
In this section, we build a classifier using the three genes with IDs (“CDKN2A|1029”, “TNFAIP6|7130”, “FABP6|2172”) to predict the groups of the samples.
Using the original kirc_DeSeq object, we apply the rlogtransformation and filter the data to the three gene: “CDKN2A|1029”, “TNFAIP6|7130”, “FABP6|2172”.
three_genes = c("CDKN2A|1029", "TNFAIP6|7130", "FABP6|2172")
kicr_log <- rlogTransformation(kirc_deseq)
kicr_r_log <- assay(kicr_log)[,]
#easier for me to handle dataframes...
kicr_r_log2 <- data.frame(assay(kicr_log))
sample_train = subset(kicr_r_log2, rownames(kicr_r_log2) %in% three_genes)Preprocessed the data:
# create annotation again for the sample data set
annotation <- data.frame(condition = rep(c("control", "treated"), each = 20))
tps_sample_train <- t(sample_train)
tps_sample_train <- cbind(tps_sample_train,annotation)
tps_sample_train$condition <- factor(tps_sample_train$condition)The sample should look like:
tps_sample_trainNext, we use lda to classify the samples into control or treated using the same samples again:
kirc_lda = lda(condition ~ ., data =tps_sample_train)
round(kirc_lda$scaling, 1)## LD1
## `CDKN2A|1029` 1.2
## `FABP6|2172` 0.0
## `TNFAIP6|7130` 0.4ghat=predict(kirc_lda)$class
table(ghat,tps_sample_train$condition)##
## ghat control treated
## control 20 1
## treated 0 19mean(ghat!=tps_sample_train$condition)## [1] 0.025As shown, there is one misclassification in the predict where it was classified as a control group instead of the actual group (treated). Overall, the classification model predicts the group effectively. However, the samples used to test the training model was the training sample.